Beta-2-adrenergic receptor (B2AR) membrane protein simulation with CHARMM¶
Use CHARMM-GUI to set up a membrane protein simulation for OpenMM.
Files¶
The initial membrane protein PDB file apo_snapshot.pdb comes from reference structures provided in Kohloff et.al. 2014, originally from PDB ID: 3POG.
A tarball of the CHARMM-GUI output can be found in charmm-gui.zip
Using CHARMM-GUI to embed B2AR in a solvated membrane¶
Go to CHARMM-GUI’s membrane builder and upload the pdb file. (
apo_snapshot.pdb). Select PDB format and clickNext Step.Stick with the default selection (Protein) and click
Next Step.Select
Terminal group patchingand select terminNTERandCTER.Select
Disulfide bondsfor the pairs184-190and106-191and clickNext Step.Select
Align the First Principal Axis Along Z(the suggestion for a homo-oligomer) and clickNext Step.Keep the default selections for
Heterogeneous Lipid,Rectangular Box Type,17.5 water thickness. SelectNumber of lipid componentsand selectPOPClipids with55lipids on the upperleaflet and lowerleaflet. ClickNext Step.Keep all default selections (
Replacement method,check lipid ring (and protein surface) penetration,Include Ions 0.15 KClandIon Placing Method: Monte-Carlo. ClickNext Step.Click
Next Stepto assemble components. When you arrive atStep 5, selectOpenMMas the Input generation option. Keep all the other default options (Generate grid information for PME FFT automaticallyandNPT ensemble. ClickNext Step.
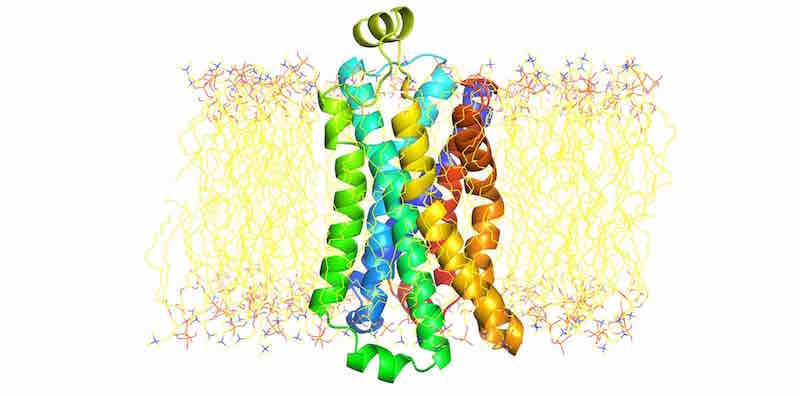
Simulating the system in OpenMM¶
The quick way¶
From the charmm-gui.tgz extract toppar/ directory and the openmm/ directory. The README file in the openmm/ directory is a csh script to run a standard equilibration and production simulation.
The flexible way¶
If you want finer control, you can write your own OpenMM code to equilibrate and simulate the system.
OpenMM can directly read CHARMM input files through the use of the openmm.app application layer. This enables the use of all the powerful setup tools in the CHARMM ecosystem that a user might be familiar with such as the CHARMM-GUI, VMD, CGenFF program
(DOI:10.1002/jcc.21367), etc. This allows users who are already working in the CHARMM environment to harness the GPU speeds that OpenMM provides without having to modify their simulation system description files.
OpenMM can also read CHARMM force field files making it is possible to use force fields that aren’t already included in OpenMM such as the general CHARMM force field (CGenFF). For example, a user can generate an str file with the CGenFF program for a ligand and load it into OpenMM. However, when using this feature, the CharmmParameterSet class needs to be used instead of the ForceField class to load all the other CHARMM force field files as
demonstrated in the example in the code below.
The example demonstrates how to use CHARMMM files that were generated with the CHARMM-GUI in an OpenMM script. The OpenMM app layer includes several classes to load CHARMM files. The CharmmPsfFile class reads the psf file and instantiates a chemical structure on which one can then call the createSystem() method to creates an OpenMM system object. For the atomic coordinates, a regular pdb file can be used or the CharmmCrdFile or CharmmRstFile classes can be used to read CHARMM
coordinate files. Files containing force field definitions come in a variety of formats such as prm, par, top, rtf, inp and str. These files are loaded into a CharmmParameterSet object which is then included as the first parameter when createSystem() is called on the chemical structure. For this example, the membrane builder in the CHARMM-GUI was used to generate the input files for the B2AR in a POPC lipid
membrane. The membrane builder provides a straightforward way to go from the RCSB X-ray structure to the protein embedded in a membrane with all the relevant CHARMM input files.
[ ]:
# download and extract the files
!wget https://openmm.org/tutorials_/b2ar_membrane/files/charmm-gui.zip
!tar xvf charmm-gui.zip
[ ]:
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
# Load CHARMM files
psf = CharmmPsfFile('charmm-gui/openmm/step5_charmm2omm.psf')
pdb = PDBFile('charmm-gui/openmm/step5_charmm2omm.pdb')
params = CharmmParameterSet('charmm-gui/toppar/par_all36_prot.prm', 'charmm-gui/toppar/top_all36_prot.rtf',
'charmm-gui/toppar/par_all36_lipid.prm', 'charmm-gui/toppar/top_all36_lipid.rtf',
'charmm-gui/toppar/toppar_water_ions.str')
# Create an openmm system by calling createSystem on psf
system = psf.createSystem(params, nonbondedMethod=CutoffNonPeriodic,
nonbondedCutoff=1*nanometer, constraints=HBonds)
integrator = LangevinIntegrator(300*kelvin, # Temperature of head bath
1/picosecond, # Friction coefficient
0.002*picoseconds) # Time step
simulation = Simulation(psf.topology, system, integrator)
simulation.context.setPositions(pdb.positions)
print("Minimizing...")
simulation.minimizeEnergy()
# Set up the reporters to report energies every 1000 steps.
simulation.reporters.append(PDBReporter('output.pdb', 1000))
simulation.reporters.append(StateDataReporter(stdout, 1000, step=True,
potentialEnergy=True, temperature=True))
# run simulation
simulation.step(10000)