Histone methyltransferase simulation with multisite Zn2+ structural ions¶
Simulating a protein with structural zinc ions using multisite ions.
This tutorial is described in OpenMM 7 publication.
Files¶
All input files can be found in hkmt_zinc.zip
Introduction¶
OpenMM allows users to model their systems using Amber and provide prmtop, and inpcrd files as input. This allows users more familiar with the Amber modeling environment to continue using their setup tools, while harnessing the speed and versatility of OpenMM. This also allows the use of non-standard force fields that have been published for use with Amber, such as metal ion models where dummy atoms are applied to mimic particular coordination geometries. Furthermore, this is facilitated
by OpenMM’s support for Extra Particles which are particles that are not ordinary atoms, such as these metal dummy atoms, virtual sites in many water models, etc.
This example illustrates the use of tleap from AmberTools to set up a simulation of the histone methyltransferase SETD2 (Uniprot: Q9BYW2), including three structural Zn2+ cations described with the tetrahedral Cationic Dummy Atom Approach (CADA: DOI:10.1007/s008940050119, Pang
lab).
We begin from the 4H12 PDB file, add missing residues (only those in the middle of the chain) and missing heavy atoms using PDBFixer, and remove unwanted water and ligand residues using MDTraj. The CADA model requires particular naming of those residues, as well as deprotonation of the ligating cysteine sulfurs (i.e. their names are changed from CYS to CYM). Finally,
tleap is run to add hydrogens, the Zn2+ dummy atoms and parametrize. We use the ff99SBildn force field and znb.lib, and frcmod.zinc files from the CADA model (downloaded from Pang lab). The prmtop and inpcrd files are saved for simulation in OpenMM.
[ ]:
# Download and unzip the inputfiles
!wget https://openmm.org/tutorials_/hkmt_zinc/files/hkmt_zinc.zip
!unzip -o hkmt_zinc.zip
[ ]:
from pdbfixer import PDBFixer
from openmm.app import PDBFile
import mdtraj as md
import os
# clean up the original PDB file and add missing residues and heavy atoms
fixer = PDBFixer('pdb4h12.ent')
fixer.findMissingResidues()
# only add missing residues in the middle of the chain, do not add terminal ones
chains = list(fixer.topology.chains())
keys = fixer.missingResidues.keys()
missingResidues = dict()
for key in keys:
chain = chains[key[0]]
if not (key[1] == 0 or key[1] == len(list(chain.residues()))):
missingResidues[key] = fixer.missingResidues[key]
fixer.missingResidues = missingResidues
fixer.findMissingAtoms()
fixer.addMissingAtoms()
PDBFile.writeFile(fixer.topology, fixer.positions, open('4h12_fixed.pdb', 'w'))
# keep only protein and zinc ions
traj = md.load('4h12_fixed.pdb')
traj = traj.atom_slice(traj.top.select('(protein and not resname SAH) or resname ZN'))
# implement changes necessary for the use of the dummy atom Zn2+ model
# change residue name of the zincs from ZN to ZNB, and atom names from ZN to Zn
for residue in traj.top.chain(1).residues:
residue.name = 'ZNB'
for atom in traj.top.chain(1).atoms:
atom.name = 'Zn'
# change name of cysteines coordinating zincs to CYM (deprotonated cysteine)
for residue in traj.top.chain(0).residues:
if residue.index in [86, 92, 82, 69, 54, 52, 73, 184, 233, 238, 231]:
residue.name = 'CYM'
traj.save('4h12_fixed_protein_zn_only.pdb')
# save the tleap script to file
with open('leaprc.setd2', 'w') as f:
f.write('''
source oldff/leaprc.ff99SBildn
addAtomTypes { { "DZ" "Zn" "sp3" } { "Zn" "Zn" "sp3" } }
loadOff znb.lib
loadamberparams frcmod.zinc
x = loadPdb 4h12_fixed_protein_zn_only.pdb
addIons x Cl- 0
solvateBox x TIP3PBOX 10.0
savePdb x topology.pdb
saveAmberParm x input.prmtop input.inpcrd
quit
''')
# run tleap
# if you have amber tools installed on your system you can uncomment and run this line
#os.system('tleap -f leaprc.setd2')
# we have included the output files that get produced:
# input.inpcrd and input.prmtop
We load the prmtop and inpcrd files in, by creating AmberPrmtopFile and AmberInpcrdFile objects. Next, the System is created by calling the createSystem() method on the AmberPrmtopFile object.
Next, the LangevinIntegrator and the Simulation are set up, using the topology from the AmberPrmtopFile and positions from the AmberInpcrdFile. In this example we will use the CUDA platform, with mixed precision. The simulation is energy minimized and equilibrated for 100 steps. Reporters are attached and the production simulation propagated for 50 ns.
[ ]:
from openmm import app
import openmm as mm
from openmm import unit
from sys import stdout
# load in Amber input files
prmtop = app.AmberPrmtopFile('input.prmtop')
inpcrd = app.AmberInpcrdFile('input.inpcrd')
# prepare system and integrator
system = prmtop.createSystem(nonbondedMethod=app.PME,
nonbondedCutoff=1.0*unit.nanometers, constraints=app.HBonds, rigidWater=True,
ewaldErrorTolerance=0.0005)
integrator = mm.LangevinIntegrator(300*unit.kelvin, 1.0/unit.picoseconds,
2.0*unit.femtoseconds)
integrator.setConstraintTolerance(0.00001)
simulation = app.Simulation(prmtop.topology, system, integrator)
#properties)
simulation.context.setPositions(inpcrd.positions)
# minimize
print('Minimizing...')
simulation.minimizeEnergy()
# equilibrate for 100 steps
simulation.context.setVelocitiesToTemperature(300*unit.kelvin)
print('Equilibrating...')
simulation.step(100)
# append reporters
simulation.reporters.append(app.DCDReporter('trajectory.dcd', 1000))
simulation.reporters.append(app.StateDataReporter(stdout, 1000, step=True,
potentialEnergy=True, temperature=True, progress=True, remainingTime=True,
speed=True, totalSteps=25000000, separator='\t'))
# run 50 ns of production simulation
print('Running Production...')
simulation.step(25000000)
print('Done!')
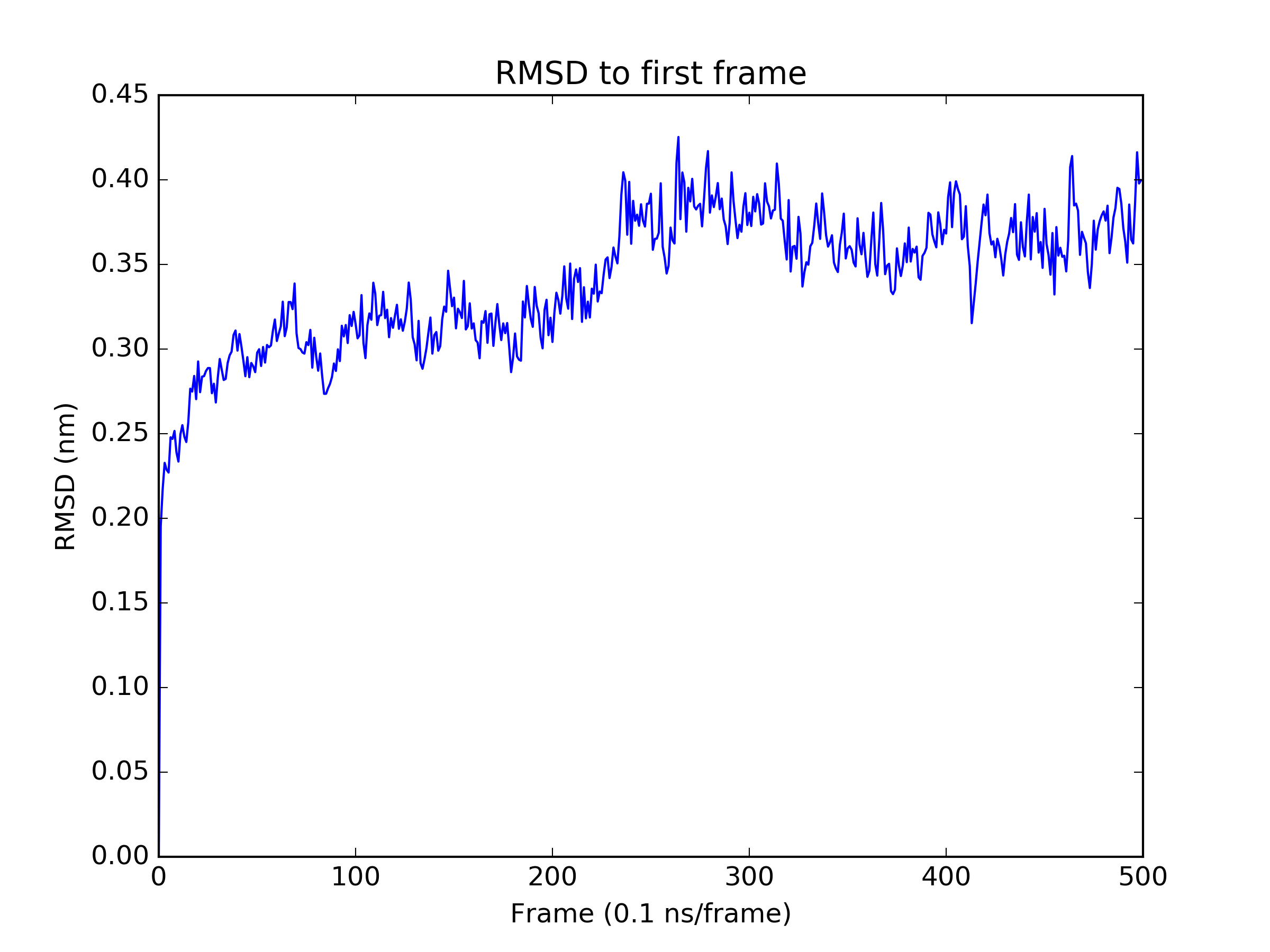
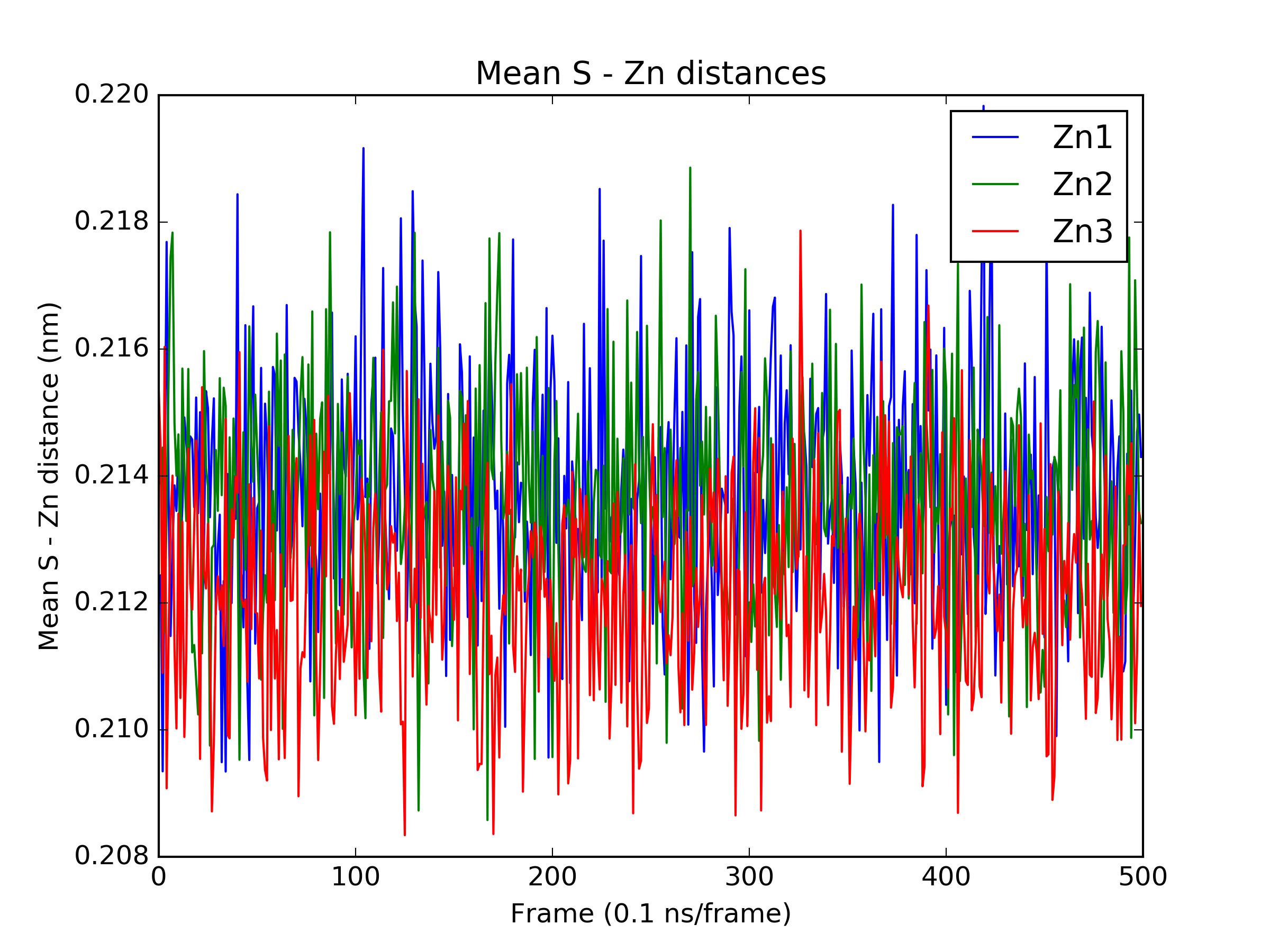
Finally, we perform a quick analysis of the trajectory. We examine the RMSD of all frames to the first frame, and the mean sulfur - zinc distances to check the stability of the metal centers.
[ ]:
import mdtraj as md
import matplotlib
matplotlib.use('Agg')
import matplotlib.pyplot as plt
import numpy as np
# load trajectory and remove solvent
traj = md.load_dcd('trajectory.dcd', top='topology.pdb', stride=50)
traj = traj.atom_slice(traj.top.select('protein or resname ZNB'))
# calculate RMSD to first frame and plot figure
rmsd = md.rmsd(traj, traj)
plt.figure()
plt.plot(rmsd)
plt.title('RMSD to first frame')
plt.xlabel('Frame (0.1 ns/frame)')
plt.ylabel('RMSD (nm)')
plt.savefig('rmsd.png', dpi=300)
plt.close()
# calculate mean sulfur - zinc distances for 3 metal centers and plot figure
atom_pairs_1 = [[3904, 892], [3904, 917], [3904, 1136], [3904, 1180]]
atom_pairs_2 = [[3909, 1136], [3909, 1336], [3909, 1392], [3909, 1470]]
atom_pairs_3 = [[3914, 2982], [3914, 3733], [3914, 3763], [3914, 3815]]
distances_1 = md.compute_distances(traj, atom_pairs_1)
distances_1 = [np.mean(x) for x in distances_1]
distances_2 = md.compute_distances(traj, atom_pairs_2)
distances_2 = [np.mean(x) for x in distances_2]
distances_3 = md.compute_distances(traj, atom_pairs_3)
distances_3 = [np.mean(x) for x in distances_3]
plt.figure()
plt.plot(distances_1, label='Zn1')
plt.plot(distances_2, label='Zn2')
plt.plot(distances_3, label='Zn3')
plt.title('Mean S - Zn distances')
plt.xlabel('Frame (0.1 ns/frame)')
plt.ylabel('Mean S - Zn distance (nm)')
plt.legend()
plt.savefig('zn_s_distances.png', dpi=300)
plt.close()